
CENTRO PROVINCIAL DE GENÉTICA MÉDICA
HOLGUÍN
Pseudoacondroplasia en una familia
Pseudoacondroplasya. Presentation of an affected family
Elayne Esther Santana HernándezI, Víctor Jesús Tamayo ChangII.
RESUMEN
Introducción:
la pseudoacondroplasia es una displasia esquelética de
transmisión autosómica dominante. Por tanto, el progenitor
afectado tiene alto riesgo de transmitir el gen mutado a su descendencia.
Los enfermos se caracterizan por una baja estatura desproporcionada,
afectaciones en los huesos largos, la columna vertebral y las
articulaciones. Debido al deficitario crecimiento de las epífisis, los
afectados padecen artrosis temprana.
Objetivo:
presentar una familia afectada de pseudoacondroplasia, de interés
debido a los pocos casos documentados en el país.
Presentación de caso:
familia con baja talla desproporcionada y alteraciones articulares, uno de
cuyos miembros -menor de 12 años- asistió por ese motivo,
acompañado de su madre, a la consulta de genética clínica.
Como antecedentes patológicos familiares se refirieron baja talla del
abuelo y los tíos maternos. Durante el examen físico del menor se
constató que su talla (por debajo del tercer percentil) y su peso
(entre el tercer y décimo percentiles), estaban en desproporción
con su edad. Presentó alteraciones en manos y pies, y escoliosis
torácica con cifosis. La madre del menor, también de baja talla,
presentó deformidades en las articulaciones de los codos y rodillas,
asociadas a dolores, y deformidad en el tórax (causada por escoliosis
de la columna torácica).
Conclusiones:
la pseudoacondroplasia es una de las displasias óseas cuyo
diagnóstico se dificulta en ocasiones por la expresión variable
del fenotipo, que en muchos casos se interpreta como baja talla familiar.
Una vez diagnosticada la enfermedad se debe asesorar a las familias
afectadas acerca del riesgo de recurrencia en la descendencia.
Palabras clave:
OSTEOCONDRODISPLASIAS/etiología,
OSTEOCONDRODISPLASIAS/fisiopatología, DEFORMIDADES CONGÉNITAS DE
LA MANO, DEFORMIDADES CONGÉNITAS DEL PIE, GENU VALGUM, ESCOLIOSIS,
LORDOSIS, CONDROCITOS, PROTEÍNAS MATRILINAS, INFORMES DE CASOS.
ABSTRACT
Introduction:
pseudoachondroplasia is a skeletal dysplasia of autosomal dominant
transmission. Therefore, the affected parent has a high risk of
transmitting the mutated gene to their offspring. The patients are
characterized by a disproportionate short stature, affectations in the long
bones, the spine and the joints. Due to the deficient growth of the
epiphyses, those affected suffer from early joint diseases.
Objective:
to present a family affected by pseudoachondroplasia, of interest due to
the few cases documented in the country.
Case presentation:
family with disproportionate short stature and joint alterations, one of
whose members -child under 12 years old- attended for that reason,
accompanied by his mother, to the clinical genetic consultation. As a
family pathological background they referred to the short stature of the
grandfather and the maternal uncles. During the physical examination of the
child it was found that his height (below the third percentile) and his
weight (between the third and tenth percentiles), were in disproportion
with his age. He presented alterations in the hands and feet, and thoracic
scoliosis with kyphosis. The mother of the child, also of low stature,
presented deformities in the joints of the elbows and knees, associated
with pain, and deformity in the thorax (caused by scoliosis of the thoracic
spine).
Conclusions:
pseudoacondroplasia is one of the bone dysplasias whose diagnosis is
sometimes difficult due to the variable expression of the phenotype, which
in many cases is interpreted as low family size. Once the disease has been
diagnosed, the affected families should be advised about the risk of
recurrence in the offspring.
Keywords:
OSTEOCHONDRODYSPLASIAS/etiology, OSTEOCHONDRODYSPLASIAS/physiopathology,
CONGENITAL HAND DEFORMITIES, CONGENITAL FOOT DEFORMITIES, GENU VALGUM,
SCOLIOSIS, LORDOSIS, CHONDROCYTES, MATRILIN PROTEINS, CASE REPORTS.
INTRODUCCIÓN
La pseudoacondroplasia es una enfermedad hereditaria, consistente en una
displasia esquelética. Fue descrita por primera vez en 1959, por
Maroteaux y Lamy. Los enfermos se caracterizan por tener baja estatura
desproporcionada, afectaciones en los huesos largos, la columna vertebral y
las articulaciones. Debido al deficitario crecimiento de las epífisis,
los afectados padecen artrosis temprana.(1)
Aproximadamente a los dos años de vida aparecen signos sospechosos de la enfermedad: desaceleración del crecimiento lineal y dificultades en la marcha. Al aumentar la edad se evidencian más la baja estatura, y las manos y pies cortos y anchos. Aunque la laxitud articular es una manifestación generalizada, afecta principalmente a las manos. Otros hallazgos físicos son braquidactilia e inclinación en genu valgum (la que provoca deformidad de las extremidades inferiores), asociada a lordosis lumbar exagerada. La deformación de las extremidades se origina por las lesiones metafisiarias.(2,3)
La pseudoacondroplasia es de transmisión autosómica dominante. Por tanto, el progenitor afectado tiene alto riesgo de transmitir el gen mutado a su descendencia. El gen responsable de la trasmisión es el COMP (19p13.1), localizado en el cromosoma 19p13.1. Este gen codifica la proteína oligomérica de la matriz del cartílago. Las mutaciones puntuales o deleciones del COMP (19p13.1) ocasionan las formas leves y graves de la enfermedad, aun cuando puedan presentarse casos aislados producto de mutaciones nuevas (de novo).(3-5)
La proteína COMP se encuentra normalmente en los espacios entre las células generadoras de cartílago (condrocitos), donde interactúa con otras proteínas. Las mutaciones del gen COMP (19p13.1) provocan la producción de proteína COMP anormal, que no puede ser transportada fuera de la célula y se acumula en los condrocitos. El efecto citotóxico de la proteína anormal causa la muerte prematura de los condrocitos durante el crecimiento óseo lineal, produciendo huesos largos acortados.(6)
En la práctica médica es frecuente encontrar pacientes de baja talla, con familiares que presentan los mismos signos clínicos, y no han sido evaluados desde el punto de vista genético. No obstante, pudiera pensarse que esta enfermedad no es frecuente debido a los pocos casos documentados en el país. De ahí que el objetivo de este trabajo sea presentar el caso de una familia afectada de pseudoacondroplasia.
PRESENTACIÓN DEL CASO
Familia con baja talla desproporcionada y alteraciones articulares, uno de
cuyos miembros -menor de 12 años- asistió a la consulta de
genética clínica por su baja talla. El paciente fue
acompañado de su madre, también de baja talla y con deformidades
en las articulaciones asociadas a dolores, principalmente en los codos y
rodillas.
Como antecedentes patológicos familiares se refirieron baja talla del abuelo y los tíos maternos. Respecto del menor, no refirió antecedentes prenatales de interés y los perinatales fueron: ruptura prematura de membranas a las 36 semanas de gestación, sufrimiento fetal agudo, parto distócico por cesárea, peso al nacer de 2 340 g, talla 47 cm y circunferencia cefálica de 31,5 cm.
Respecto del desarrollo pondoestatural y psicomotor en el primer año de vida se estableció que el niño sostuvo la cabeza a los tres meses, con control cefálico a los cuatro meses, fue capaz de sentarse a los siete meses, caminó y gateó con apoyo a los 11 meses y a los 16 caminó por sí solo. A los dos años presentó buen aumento de peso y decía monosílabos y oraciones cortas.
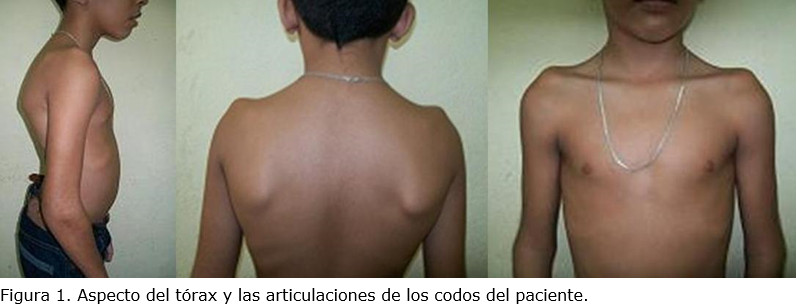
Durante el examen físico del menor se constató una talla en relación a la edad de 128 cm (por debajo del tercer percentil). Igualmente pesó 25 kg (entre el tercer y décimo percentiles) en desproporción con su edad. En los miembros superiores se destacó la limitación en la extensión de las articulaciones de los codos (semiflexión), y deformidad de la columna vertebral consistente en escoliosis torácica con cifosis (figura 1).
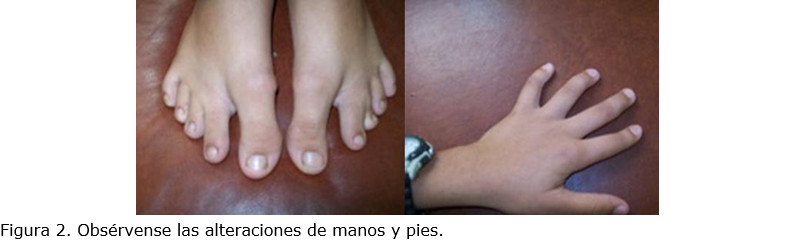
Los pies, pequeños y anchos, con dedos cortos, excepto el primer dedo grueso, separado del resto, y presencia de un surco profundo entre el primer y segundo dedos. Las manos, también pequeñas, con hiperlaxitud articular y clinodactilia del quinto dedo (figura 2).
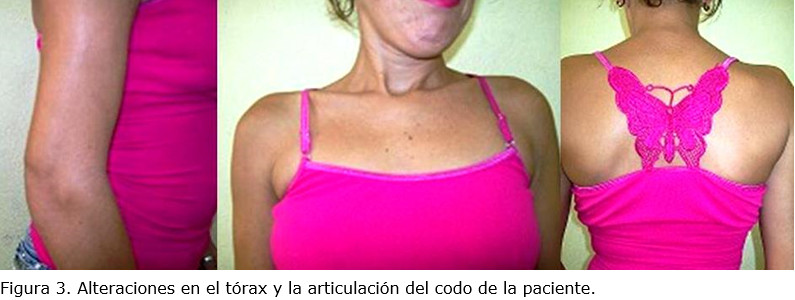
La madre del menor, de estatura baja (137 cm), presentó semiflexión de los codos, deformidad en el tórax (causada por escoliosis de la columna torácica) y dolores articulares relacionados con la deformidad de las articulaciones (figura 3).
Con el consentimiento informado de los miembros de la familia, se les tomaron fotografías ilustrativas del caso para su publicación. Para ello se cumplieron estrictamente los principios éticos de la investigación médica, y se tuvo cuidado de no revelar la identidad de los pacientes.
DISCUSIÓN
La pseudoacondroplasia una deficiencia grave del crecimiento,
acompañada de deformidades esqueléticas tales como piernas
arqueadas e hiperlordosis.(1) Aunque se plantea que es un
trastorno poco frecuente, con una prevalencia estimada de aproximadamente
1/30 000 enfermos,(2) la prevalencia al nacimiento no se conoce
porque los recién nacidos afectados son indistinguibles de los sanos.(1-3)
Los signos clínicos de la pseudoacondroplasia son múltiples: brazos y piernas cortos, demoras en gatear y caminar, marcha ondulante, dolor en las articulaciones a edad temprana, limitación del movimiento en los codos y caderas, rango anormal de movimiento articular (hiperextención) en las manos, rodillas y tobillos, deformidades de la rodilla, deformidades de la columna vertebral (escoliosis, lordosis y curvatura hacia dentro de las porciones cervical y lumbar de la columna vertebral).(4)
Esta afección genética se diagnostica a partir del estudio radiológico de las anomalías epifisarias y metafisarias que presentan los enfermos. Las anomalías consisten en epífisis irregulares con forma de hongo, aplanamiento de las vértebras, aspecto craneofacial normal, huesos largos con metafisis amplias y anormalidades en las vértebras (causadas por el grado variable de aplanamiento).(5) Los pacientes presentan hipocrecimiento posnatal asociado a hipermovilidad de las articulaciones mayores, excepto del codo, con desviación cubital de la mano y dedos cortos hipermovibles.(6)
Desde el punto de vista genético todas las mutaciones del gen COMP (19p13.1) son estructurales en los exones 8-14 que codifican los dominios de unión al calcio, o son repeticiones tipo 3 y 14-19 del dominio globular carboxilo terminal. Aproximadamente 30 % de los individuos con pseudoacondroplasia tienen la misma mutación recurrente: la deleción de un triplete GAC (p.Asp473 del c.1417_1419 del GAC) dentro de una secuencia de cinco codones GAC en el exón 13.(6-8)
El diagnóstico diferencial se debe hacer respecto de otras displasias óseas como la acondroplasia y la hipoacondroplasia. La primera se manifiesta con anomalías craneofaciales, baja talla a expensas del acortamiento rizomérico de húmero y el fémur, y difiere de la pseudoacondroplasia en las manifestaciones radiológicas. Los enfermos de hipoacondroplasia no presentan alteraciones craneofaciales, pero manifiestan discapacidad intelectual ligera.(9)
En investigaciones realizadas en familias con baja talla desproporcionada como único signo, también se ha detectado la mutación en el gen COMP. La variabilidad de la expresión fenotípica pudiera ser la causa de los pocos casos registrados.(10)
CONCLUSIONES
La pseudoacondroplasia es una de las displasias óseas cuyo
diagnóstico se dificulta en ocasiones por la expresión variable
del fenotipo, que en muchos casos se interpreta como baja talla familiar.
Una vez diagnosticada la enfermedad se debe asesorar a las familias
afectadas acerca del riesgo de recurrencia en la descendencia.
REFERENCIAS BIBLIOGRÁFICAS
Recibido: 30 de enero de 2017
Aprobado: 16 de mayo de 2017
| MsC. Elayne Esther Santana Hernández Centro Provincial de Genética Médica Avenida de los Libertadores No.91, Reparto Peralta. Holguín, Cuba. CP.80100 Correo electrónico: elsantana@infomed.sld.cu |