Publicación Científica de la Universidad de Ciencias Médicas de Ciego de Ávila Editorial Ciencias Médicas |
 |
REVISTA MÉDICA ELECTRÓNICA DE
CIEGO DE ÁVILA |
2022;28:e2916
ISSN: 1029-3035
RNPS: 1821 |
|

Informe de caso
Atención anestésica de la enfermedad de Steiner. Informe de caso
Anesthetic care of Steiner's disease. case report
Alejandro Valdés-Torres1* https://orcid.org/0000-0002-6525-1762
Shemanet García-Cid2 https://orcid.org/0000-0001-7199-4696
Norys Quintana-Santos3 https://orcid.org/0000-0001-7600-6749
1Especialista en Anestesiología y Reanimación. Profesor Instructor Hospital General Provincial Docente “Cptán Roberto Rodríguez Fernández” Morón. Ciego de Ávila, Cuba.
2Especialista en Segundo Grado en Anestesiología y Reanimación y de Primer Grado en Medicina General Integral. Diplomada en Cuidados Intensivos. Profesora Instructora. Hospital General Provincial Docente “Cptán Roberto Rodríguez Fernández” Morón. Ciego de Ávila, Cuba.
3Máster en Urgencias y Emergencias Médicas. Especialista de Primer Grado en Anestesiología y Reanimación. Profesor Instructor. Hospital General Provincial Docente “Cptán Roberto Rodríguez Fernández” Morón. Ciego de Ávila, Cuba.
* Autor para la correspondencia. Correo electrónico: alexvtcu@gmail.com
RESUMEN
Introducción: la distrofia miotónica tipo 1 o enfermedad de Steinert (OMIM 160900, ORPHA 273, CIE-9-MC359.21, CIE-10 G71.1, CIE-11 8C71.0) es la miopatía de mayor prevalencia en el adulto, aunque existen dos formas de presentación pediátrica: la distrofia miotónica congénita y la variante de inicio infantil. Constituye un reto para el anestesiólogo por las complicaciones que pueden presentarse durante el acto quirúrgico.
Objetivo: describir la actuación anestésica en un paciente con enfermedad miotónica de Steiner.
Presentación del caso: paciente de 11 años de edad y 58 Kg de peso, blanco, masculino, que es anunciado para exploración laparoscópica por testículo derecho no descendido. Con antecedentes de parto distócico demorado y cianosis al nacer, que requirió ventilación artificial en neonatología por período de tres días, con retraso del desarrollo psicomotor al que se le diagnostica durante el período de lactante, la enfermedad miotónica de Steiner. Se describe la técnica anestésica utilizada, los fármacos y procedimientos de control en el preoperatorio, durante el acto quirúrgico y en el período de recuperación.
Conclusiones: la enfermedad de Steiner es un reto para el anestesiólogo por los efectos multisistémicos que pueden conducir a complicaciones perioperatorias e incluso a la muerte durante el acto quirúrgico o la recuperación anestésica. La conducta terapéutica debe ser individualizada y valorada durante el perioperatorio, se recomienda una planificación anestésica minuciosa, anticipada a las complicaciones.
Palabras clave: DISTROFIA MIOTÓNICA; DISTROFIAS MUSCULARES; ANESTESIA ENDOTRAQUEAL; ATENCIÓN PERIOPERATIVA; NIÑO
ABSTRACT
Introduction: myotonic dystrophy type 1 or Steinert's disease (OMIM 160900, ORPHA 273, ICD-9-MC359.21, ICD-10 G71.1, ICD-11 8C71.0) is the most prevalent myopathy in adults, although there are two forms of pediatric presentation: congenital myotonic dystrophy and the infantile onset variant. It constitutes a challenge for the anesthesiologist due to the complications that may arise during the surgical act.
Objective: to describe the anesthetic action in a patient with Steiner's myotonic disease.
Case presentation: 11-year-old white male patient weighing 58 kg, who was announced for laparoscopic exploration for undescended right testis. With a history of delayed dystocic delivery and cyanosis at birth, which required artificial ventilation in neonatology for a period of three days, with delayed psychomotor development diagnosed during the infant period, Steiner's myotonic disease. The anesthetic technique used, the drugs and control procedures in the preoperative period, during the surgical act and in the recovery period are described.
Conclusions: Steiner's disease is a challenge for the anesthesiologist due to the multisystemic effects that can lead to perioperative complications and even death during the surgical procedure or anesthetic recovery. Therapeutic conduct must be individualized and assessed during the perioperative period, a thorough anesthetic planning is recommended, anticipating complications.
Keywords: MYOTONIC DYSTROPHY; MUSCULAR DYSTROPHIES; ANESTHESIA, ENDOTRACHEAL; PERIOPERATIVE CARE; CHILD
Recibido: 13/11/2020
Aprobado: 15/02/2021
INTRODUCCIÓN
La distrofia miotónica tipo 1, también llamada enfermedad de Steinert, enfermedad de Steinert-Curschmann, enfermedad de Batten-Gibb y miotonía atrófica (OMIM 160900, ORPHA 273, CIE-9-MC359.21, CIE-10 G71.1, CIE-11 8C71.0), es la miopatía de mayor prevalencia en el adulto.(1,2) Esta enfermedad presenta un patrón de herencia autosómico dominante y se produce por la expansión de tripletes citosina-timina-guanina (CTG) en la región no codificante del gen DMPK, localizado en el brazo largo del cromosoma 19 (19q13.3); en ella son características las alteraciones neuromusculares, pero produce también alteraciones sistémicas.(1)
Es considerada una de las enfermedades con mayor variedad fenotípica, lo que conlleva a una necesidad de tratamiento personalizado de cada paciente, así como un conocimiento adecuado y profundo de las alteraciones de cada órgano y sistema, para poder ofrecer a los pacientes el seguimiento y tratamiento más apropiado.(1)
La distrofia miotónica tipo 1 se manifiesta habitualmente en la edad adulta, pero existen dos formas de presentación pediátrica: la distrofia miotónica congénita y la variante de inicio infantil. Ambas tienen características diferentes respecto a la forma clásica del adulto, aunque el proceso diagnóstico es similar: la sospecha clínica seguida de la confirmación genética mediante el estudio dirigido.
La distrofia miotónica congénita, es la forma más grave, pero también la más infrecuente. La incidencia varía entre 2,1 y 5,2 por 100.000 recién nacidos vivos.(3,4) En España se ha estimado en 0,8 por 10.000.(5)
En la mayoría de los casos la transmisión es por vía materna. Las manifestaciones de la enfermedad están presentes desde el nacimiento. De acuerdo con Moxley y cols.,(6) los antecedentes obstétricos suelen reflejar la escasa movilidad del feto en el período prenatal: movimientos fetales reducidos, polihidramnios y deformidades articulares detectados mediante ecografía, o una gestación finalizada mediante cesárea debido a que el parto no progresa. Las dificultades en el período expulsivo suponen un riesgo añadido de asfixia perinatal.(6)
El neonato muestra al nacer hipotonía marcada asociada a debilidad y pobreza de movimientos. La facies es característica, con paresia facial y labio superior en forma de V invertida. Es muy común la presencia de problemas respiratorios (más del 50 % de los casos) por debilidad del diafragma y de los músculos intercostales, inmadurez pulmonar (el riesgo de prematuridad está aumentado) y fallo del control respiratorio cerebral. También existen dificultades en la succión-deglución, debidas a la debilidad de la musculatura bulbar.(7) Las retracciones articulares son habituales, especialmente en forma de pie zambo o equino-varo. Al contrario de lo que sucede con el adulto, los problemas cardiovasculares no son frecuentes a esta edad y no existe miotonía clínica ni eléctrica hasta edades posteriores, por lo que no está indicada la realización de electromiograma. Se ha descrito una mayor incidencia de ventriculomegalia y de malformaciones del desarrollo cortical cerebral.(8)
El diagnóstico diferencial se establece preferentemente con las distrofias musculares y miopatías congénitas y con los síndromes miasteniformes congénitos.
Los niños que sobreviven al sexto mes de vida mejoran lentamente, aunque manifiestan un retraso evidente en las habilidades motoras. Prácticamente todos consiguen la marcha autónoma y hacia los tres o cuatro años mejora la hipotonía y las dificultades para la alimentación. La prominente debilidad facial se mantiene, lo que ocasiona la típica “boca de carpa”.
La miotonía no aparece hasta la adolescencia. Frente a los progresos motores, en los primeros años se pone de manifiesto una afectación cognitiva de intensidad variable, aunque existe una minoría de casos con capacidad intelectual normal.(9) El cociente de inteligencia se sitúa de media en 70, pero el retraso mental puede llegar a un grado moderado o grave. Este es uno de los principales problemas de la forma congénita. Los niños pueden presentar rasgos del espectro autista y otros problemas conductuales asociados.(10) El aspecto facial puede condicionar que al niño se le atribuya una capacidad cognitiva menor de la que realmente tiene. El desarrollo del lenguaje puede estar interferido, tanto por la debilidad en la musculatura de la boca, paladar y mandíbula, que dificultan una buena pronunciación, como por el riesgo de hipoacusia ante las infecciones repetidas del tracto respiratorio superior. Con los años aparece la debilidad en extremidades con distribución distal, así como el resto de las manifestaciones propias de la edad adulta.
Los pacientes con distrofia miotónica tipo 1 tienen algunos riesgos específicos cuando son sometidos a procedimientos anestésicos, que deben tenerse en consideración. Son frecuentes en estos pacientes, intervenciones quirúrgicas para el tratamiento de las cataratas e intervención quirúrgica abdominal, sobre todo la colecistectomía, además de las posibles intervenciones por otros trastornos. La anestesia locorregional es segura, pero los opiáceos y sedantes con efecto respiratorio deben utilizarse con cautela, pues estos pacientes son especialmente sensibles a estos fármacos y pueden, incluso, sufrir reacciones paradójicas con relajantes musculares. La anestesia raquídea es la más recomendable para la intervención quirúrgica tocológica.(11,12)
Debido a que su presentación es infrecuente, el manejo anestésico es complicado y a que existen en Cuba pocos casos con informes sobre la conducta a seguir, el objetivo del presente trabajo es describir la actuación anestésica en un paciente con enfermedad miotónica de Steiner.
INFORMACIÓN DEL PACIENTE
Paciente de 11 años de edad y 58 Kg de peso, blanco, masculino, que es anunciado para exploración laparoscópica por testículo derecho no descendido. Con antecedentes de parto distócico demorado y cianosis al nacer, requirió ventilación artificial en el servicio de Neonatología por período de tres días; con retraso del desarrollo psicomotor diagnosticado en la etapa de lactante como enfermedad miotónica de Steiner. Se declara apto para la intervención quirúrgica después de evaluarse por los especialistas de Neurología.
CUMPLIMIENTO DEL COMPONENTE ÉTICO DE LA INVESTIGACIÓN CLÍNICA
El comité de ética de la investigación de la institución aceptó la publicación del informe de caso, previa aprobación de la madre del paciente. Esta última se obtuvo mediante la firma del consentimiento informado para divulgar su situación de salud, que incluyó el permiso para publicar las fotos. Se eliminó la información identificativa de todos los datos relacionados con el paciente.
PERSPECTIVA DEL PACIENTE
El paciente en todo momento se mantuvo cooperativo con la atención médica recibida. Al finalizar, tanto él como sus familiares, mostraron satisfacción y agradecimiento con los resultados del tratamiento.
HALLAZGOS CLINICOS
Medicación anterior: no referida.
Antecedentes Patológicos Familiares: no referidos.
Historia anestésica: no referida.
Examen físico:
Sistema cardiovascular: ruidos cardíacos rítmicos de buen tono e intensidad, no se ausculta soplo, pulsos periféricos presentes. Tensión arterial: 100/70mmHg
Frecuencia cardiaca: 72 latidos por minuto; sistema respiratorio: murmullo vesicular audible, no se auscultan estertores. Frecuencia respiratoria: 16 respiraciones por minuto.
Fosas nasales: permeables.
Boca: Test de Mallampati: III Apertura bucal: 3cm (Figura 1)
Tráquea: central y movible.
Columna vertebral: sin alteraciones.
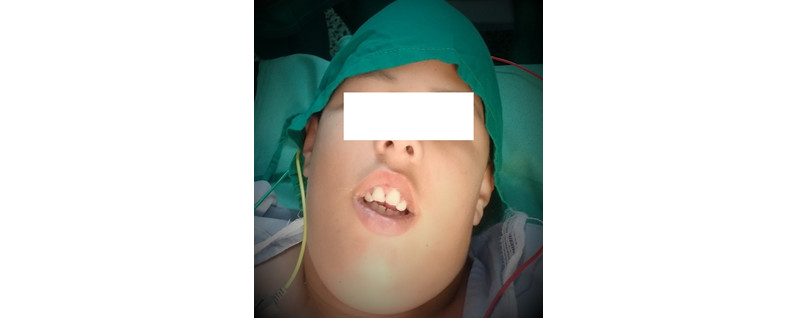
Fig. 1 - Apertura bucal
EVALUACIÓN DIAGNÓSTICA
Hematocrito: 0.49 Tiempo de coagulación: 8 s Tiempo de sangrado: 1 s
Clasificación de la Sociedad Americana de Anestesiología: ASA III.
Riesgo quirúrgico: regular.
Anestesia propuesta: general endotraqueal.
INTERVENCIÓN TERAPÉUTICA
En la unidad quirúrgica, se realizó monitorización estándar de electrocardiograma, tensión arterial no invasiva, frecuencia cardiaca, ritmo cardiaco, saturación parcial de oxígeno por pulsooximetría (SpO2), capnografía y se obtuvieron los signos vitales siguientes: tensión arterial no invasiva: 100/60 mmHg, frecuencia cardiaca: 68l latidos por minuto, SpO2: 98%, frecuencia respiratoria: 16 respiraciones por minuto y EtCO2: 38 mmHg.
En el preoperatorio inmediato se suministró ranitidina 150mg, para evitar complicaciones gastrointestinales, así como atropina 0,5mg para mejorar la frecuencia cardiaca hasta alcanzar 80 latidos por minuto. Se pre oxigenó con máscara facial y una fracción inspirada de oxígeno al 100 % durante cinco minutos.
La inducción anestésica fue con propofol 2,5 mg/Kg de peso (150 mg), fentanilo 2 mg/Kg de peso (100µg) y atracurio 0.4 mg/Kg de peso (25mg) por vía endovenosa. Previa laringoscopia se administró lidocaína 2% 1,5 mg/Kg (90 mg).
La laringoscopia fue ligeramente difícil (Cormarck y Lehane grado III-IV) (Figura 2); se intubó con tubo orotraqueal 7.0 con estilete, se comprobó ventilación mediante auscultación y se acopló a una máquina MINDRAY con circuito semicerrado y modalidad de control de volumen.

Fig. 2 - Grado de visualización de la laringoscopia (Clasificación de Cormack y Lehane grado II/IV)
El mantenimiento anestésico se realizó con fentanilo 2 µg/Kg de peso por vía endovenosa, atracurio a 0,2 mg/Kg de peso y una combinación de oxígeno, aire comprimido y halogenados en discretas concentraciones por la susceptibilidad de estos pacientes a los mismos.
Durante la intervención quirúrgica el paciente se mantuvo estable hemodinámicamente y la intervención duró 45 minutos. Solo fue necesario administrar dos dosis de fentanilo y 35 mg de atracurio. Se potenció el efecto de los gases halogenados como el sevoflurano en concentraciones de 0,6-0,2 % para no tener que usar dosis subsecuentes de relajantes musculares no despolarizantes y no empeorar la relajación muscular característica en estos pacientes.
En el transoperatorio se administró diclofenaco de sodio, 75 mg por vía endovenosa, para el control del dolor, además al concluir la cirugía se administró ondansetron 4 mg por vía endovenosa para contrarrestar los efectos de las náuseas y vómitos postoperatorios.
En la recuperación anestésica no se realizó reversión del bloqueo neuromuscular para no potenciar la bradicardia del paciente y otras complicaciones. Se obtuvo una recuperación espontánea en solo cinco minutos.
Se comprobó la recuperación del paciente, al presentar una SpO2 del 98 – 100% y un volumen de 4,5 l/min según espirómetro de Ray, despierto, consciente, estable hemodinámicamente con cifras de tensión arterial no invasiva 110/70mmHg, frecuencia cardiaca de 88 latidos por minuto, frecuencia respiratoria 15 respiraciones por minuto, con diuresis de 150 ml y sin dolor y se procedió a extubar sin complicaciones.
En el postoperatorio mediato no se encontraron evidencias de que la hipotonía hubiese empeorado, ni tampoco la función respiratoria.
DISCUSIÓN
Los cuidados perioperatorios en este tipo de pacientes son primordiales. Se recomienda desde el preoperatorio la vigilancia cardiorrespiratoria con electrocardiograma y SpO2. Según se requiera, se pueden indicar otros estudios como: pruebas funcionales respiratorias, radiografía de tórax a distancia de tele y hemogasometría. Es necesario evitar premedicar con opioides y tener precaución con las benzodiacepinas, debido a que estos pueden producir hipoxia e hipercapnia por debilidad de los músculos respiratorios. (13)
En el transoperatorio se debe considerar la posibilidad de luxación de la articulación temporomandibular, al realizar la laringoscopia por lo que se recomienda especial cuidado.
En la inducción, evitar los fármacos de metabolismo lento, se recomienda la disminución de la dosis; para la relajación muscular, utilizar bloqueadores neuromusculares no despolarizantes de corta acción en dosis pequeñas, ya que se puede prolongar la recuperación, por lo que es muy útil la monitorización neuromuscular; la neostigmina puede producir bloqueo por despolarización con acetilcolina. No se recomienda la succinilcolina debido a que esta puede desencadenar hiperpotasemia y bloqueo ganglionar.
En el momento de realizar la laringoscopia se realizará presión cricoidea (maniobra de Sellick) con el objetivo de evitar aspiración de contenido gástrico.
En el postoperatorio inmediato se recomienda mantener vigilancia estricta sobre las funciones cardiovasculares, respiratorias y neuromusculares, la analgesia adecuada con medicamentos antiinflamatorios no esteroideos o anestésicos locales, que mantuvieron niveles entre cero y tres puntos en la escala visual analógica. La complejidad del cuadro clínico, ayuda a elegir el fármaco y la dosis adecuada para este tipo de pacientes, ya que si es de reciente instalación, los efectos deletéreos de los anestésicos son menos pronunciados. (14)
El cuidado primordial es impedir el desencadenamiento de una crisis miotónica, que ocasiona aumento del consumo de oxígeno y del débito cardiaco, lo que pudiera precipitar la insuficiencia cardiorrespiratoria. Ésta puede desencadenadarse de muchas maneras: hipocalemia, miedo, ayuno prolongado, hipoxemia, hipercarbia, dolor, ansiedad, descarga adrenérgica, bisturí eléctrico, hipotermia, estimulador del nervio periférico, temblores, fármacos anticolinesterásicos, cambios en el pH, en electrolitos séricos, esfuerzo voluntario. La miotonía se puede desencadenar en el masetero, que es un músculo sensible. El tratamiento de la crisis miotónica debe ser con anestésicos locales y fármacos antiarrítmicos clase I, ya que producen bloqueo de los canales de sodio y disminuyen la hiperexcitabilidad de la membrana.(13)
El empleo de técnicas anestésicas locorregionales es el procedimiento de elección, en los pacientes con distrofia muscular de Steinert, ya que evitará la propagación del impulso nervioso y por ende de la contracción muscular, pero no hay que descuidar aspectos importantes como la temperatura, asegurándonos del adecuado aporte de glucosa. En caso de elegir administrar anestesia general, es importante ser cuidadosos al momento de la emersión y evaluar meticulosamente la extubación del paciente, así como las medidas de soporte ventilatorio que se toman en el postoperatorio.
CONCLUSIONES
La enfermedad de Steiner es un reto para el anestesiólogo por los efectos multisistémicos que pueden conducir a complicaciones perioperatorias e incluso a la muerte durante el acto quirúrgico o la recuperación anestésica. La conducta terapéutica debe ser individualizada; durante el perioperatorio se exige una planificación anestésica minuciosa para anticiparse a las complicaciones. La seguridad garantizada al paciente durante el proceso anestésico y quirúrgico permitió los resultados favorables descritos.
REFERENCIAS BIBLIOGRÁFICAS
- Gutiérrez-Gutiérrez G, Díaz-Manera J, Almendrote M, Azriel S, Barcena JE, Cabezudo-García P, et al. Guía clínica para el diagnóstico y seguimiento de la distrofia miotónica tipo 1, DM1 o enfermedad de Steinert. Med Clin (Barc) [Internet]. 2018 [citado 23 Ene 2020];153(2):[aprox 22 p.]. Disponible en: https://www.researchgate.net/profile/Gerardo-Gutierrez-Gutierrez-2/publication/332466662_Guia_clinica_para_el_diagnostico_y_seguimiento_de_la_distrofia_miotonica_tipo_1_DM1_o_enfermedad_de_Steinert/links/5d1cf1c0299bf1547c955354/Guia-clinica-para-el-diagnostico-y-seguimiento-de-la-distrofia-miotonica-tipo-1-DM1-o-enfermedad-de-Steinert.pdf
- Fernández-Torrón R, Maneiro-Vicente M, Martí-Carrera I, La fuente-Hidalgo M, Cobo-Esteban AM, Martorell-Sampol L, et al. Miotonías distróficas. En: Gutiérrez-Rivas E, editor. Manual de enfermedades neuromusculares. Maja-dahonda: Ergón; 2017. p. 417–27.
- Campbell C, Levin S, Siu VM, Venance S, Jacob P. Congenital myoto-nic dystrophy: Canadian population-based surveillance study. J Pediatr 2013;163(1):120-5.
- Darin N, Tulinius M. Neuromuscular disorders in childhood: A descriptive epi-demiological study from western Sweden. Neuromuscul Disord. 2000;10:1-9.
- González-de Dios J, Martínez-Frías ML, Egues-Jimeno J, Gairi-Tahull JM, Gómez-Sabrido F, Morales-Fernández MC, et al. Estudio epidemiológico de la distrofiamiotónica congénita de Steinert: características dismórficas. An Esp Pediatr [Internet]. 1999 [citado 23 Ene 2020];51(4):389-96. Disponible en: https://www.aeped.es/sites/default/files/anales/51-4-12.pdf
- Moxley RT, Ciafaloni E, Guntrum D. Myotonic dystrophy. En: Darras BT, Jones R, Ryan MM, de Vivo DC, editores. Neuromuscular disorders of infancy, childhood,and adolescence: A clinician’s approach. London: Elsevier; 2015. p. 697–718.
- Suáreza B, Araya G. Síndrome hipotónico como manifestación de enfermedad neuromuscular hereditaria en la infancia. Rev. med. clin. Condes [Internet]. 2018 [citado 23 Ene 2020];29(5):502-511. https://www.capacitacionesonline.com/blog/wp-content/uploads/2018/10/S%C3%ADndrome-hipot%C3%B3nico-como-manifestaci%C3%B3n-de-enfermedad-neuromuscular-hereditaria-en-la-infancia.-REV.-MED.-CLIN.-CONDES-2018.pdf
- Mutchnick IS, Thatikunta MA, Gump WC, Stewart DL, Moriarty TM. Congeni-tal myotonic dystrophy: Ventriculomegaly and shunt considerations for thepediatric neurosurgeon. Childs Nerv Syst [Internet]. 2016 [citado 23 Ene 2020];32:609–16. Disponible en: https://link.springer.com/content/pdf/10.1007/s00381-015-2993-y.pdf
- Rosado-Bartolomé A, Gutiérrez-Gutiérrez G. La distrofia miotónica de Steinerten urgencias. Emergencias [Internet]. 2016 [citado 23 Ene 2020];28(4):270–2. Disponible en: http://emergencias.portalsemes.org/descargar/la-distrofia-miotnica-de-steinert-en-urgencias/force_download/
- Seijas-Gómez R, Basterra-Jiménez I, Luna-Lario P, Tirapu-Ustarroz J, Cabada-Giadas T, Iridoy-Zulet M, et al. Estudio descriptivo del perfil neuropsicológicoy psicopatológico en pacientes con distrofia miotónica tipo 1. Rev Neurol [Internet].2015 [citado 23 Ene 2020];61(12):529-35. Disponible en: https://academica-e.unavarra.es/bitstream/handle/2454/28451/distrofia.pdf?sequence=1&isAllowed=y
- Molero-Díez YB, Sánchez-Hernando VJ, Ruiz-Simón FA. Aclaración en el manejo anestésico en la enfermedad de Steinert. Rev Mex Anestesiol [Internet]. 2021 [citado 23 Ene 2020]; 44(4):31. Disponible en: https://www.scielo.org.mx/pdf/rma/v44n4/0484-7903-rma-44-04-315.pdf
- Molero-Díez YB, Sánchez-Hernando VJ, Ruiz-Simón FA, Gómez-Fernández M, Mateos-Arribas MT, García-Lázaro F. Manejo anestésico en la enfermedad de Steinert. A propósito de un caso. Rev. mex. Anestesiol [Internet]. 2021 [citado 23 Ene 2020];44(1):66-69. Disponible en: https://www.scielo.org.mx/pdf/rma/v44n1/0484-7903-rma-44-01-66.pdf
- Oriol-López SA, Hernández-Bernal CE. Anestesia en la distrofia muscular de Steinert. Rev Mex Anest [Internet]. 2010 [citado 23 Ene 2020];33(3):160-5. Disponible en: https://anestesiar.org/WP/uploads/2012/04/cma103g.pdf
- Vilaplana BJ, Villalonga MA. Capítulo 46. Aspectos clínicos del uso de bloqueadores neuromusculares en enfermedades neuromusculares. En: Álvarez-Gómez JA, González-Miranda F, Bustamante-Bozza R. Relajantes Musculares en Anestesia y Terapia intensiva, 2ª. Ed. Madrid: Arán, 2000. p.599-612.
Conflictos de intereses
Los autores declaran que no existen conflictos de intereses.
Contribuciones de los autores
Alejandro Valdés-Torres: Propuesta de las técnicas anestésicas, confección del trabajo, aprobación de la versión final del manuscrito.
Shemanet García-Cid: ayuda en la realización de las técnicas anestésicas recolección de los datos y aprobación final del manuscrito.
Norys Quintana-Santos: recolección de los datos, revisión crítica y aprobación final del manuscrito.
Financiación
Hospital General Provincial Docente “Cptán Roberto Rodríguez Fernández”, Morón.
