Publicación Científica de la Universidad de Ciencias Médicas de Ciego de Ávila Editorial Ciencias Médicas |
 |
REVISTA MÉDICA ELECTRÓNICA DE
CIEGO DE ÁVILA |
2022;28:e2260
ISSN: 1029-3035
RNPS: 1821 |
|

Informe de caso
Colesteatoma gigante en fosa posterior. Informe de caso
Giant cholesteatoma in the posterior fossa. Case report
Ernesto Enrique Horta-Tamayo1* https://orcid.org/0000-0002-1292-1689
Carlos Alexander García-Alonso2 https://orcid.org/0000-0001-9657-6351
Luis César Acosta-González3 https://orcid.org/0000-0002-6463-4243
1Especialista de Primer Grado en Neurocirugía. Profesor Asistente. Investigador Agregado. Hospital Clínico-Quirúrgico “Lucía Iñiguez Landín”. Holguín, Cuba.
2Especialista de Primer Grado en Neurocirugía. Profesor Asistente. Hospital Clínico-Quirúrgico “Lucía Iñiguez Landín”. Holguín, Cuba.
3Especialista de Primer Grado en Neurocirugía. Hospital Clínico-Quirúrgico “Lucía Iñiguez Landín”. Holguín, Cuba.
*Autor para la correspondencia. Correo electrónico: ernestoht@infomed.sld.cu
RESUMEN
Introducción: los colesteatomas congénitos son lesiones benignas del oído medio, de crecimiento lento y progresivo; pueden alcanzar gran tamaño antes de ocasionar síntomas, que revelan su presencia solo en estadios avanzados. La intervención quirúrgica es la modalidad de tratamiento en todos los casos. No existen reportes de casos similares en Cuba.
Objetivo: presentar un caso de un paciente con las características clínicas, imagenológicas y anatomopatológicas de un colesteatoma congénito de fosa posterior.
Presentación del caso: paciente masculino de 48 años de edad, que comienza con tinnitus e hipoacusia izquierda y pérdida del equilibrio. El examen neurológico, reveló dismetría, discronometría y nistagmo horizontal. En estudios imagenológicos se informó lesión ocupativa en ángulo pontocerebeloso sin erosión ósea. El paciente rechaza la cirugía durante tres años y desarrolla nuevos síntomas dados por parestesias trigeminales, cefalea y disfonía, además de parálisis facial periférica izquierda. Se realiza abordaje retrosigmoideo suboccipital, se encontró lesión blanca perlada relacionada con colesteatoma, confirmado por biopsia. Se logró resección subtotal. Presentó meningitis química postquirúrgica la cual resolvió espontáneamente. La evolución fue satisfactoria.
Conclusiones: los colesteatomas congénitos intracraneales son lesiones muy infrecuentes y en ocasiones son similares a otras lesiones en la fosa craneal posterior. Imagenológicamente, se presentan como lesiones bien delimitadas, que no presentan realce a la administración de contraste. En este paciente la evolución fue favorable, a pesar del tratamiento quirúrgico tardío, por su negativa inicial.
Palabras clave: COLESTEATOMA DEL OÍDO MEDIO/congénito; COLESTEATOMA DEL OÍDO MEDIO/cirugía; QUISTE DERMOIDE; QUISTE EPIDÉRMICO; FOSA CRANEAL POSTERIOR; TOMOGRAFÍA COMPUTARIZADA DE EMISIÓN; IMAGEN POR RESONANCIA MAGNÉTICA; INFORMES DE CASOS.
ABSTRACT
Introduction: congenital cholesteatomas are benign lesions of the middle ear, with slow and progressive growth; They can reach a large size before causing symptoms, which reveal their presence only in advanced stages. Surgical intervention is the treatment modality in all cases. There are no reports of similar cases in Cuba.
Objective: to present a case of a patient with the clinical, imaging and pathological characteristics of a congenital cholesteatoma of the posterior fossa.
Case presentation: 48-year-old male patient, who started with tinnitus and left hearing loss and loss of balance. The neurological examination revealed dysmetria, dyschronometry and horizontal nystagmus. Imaging studies reported an occupying lesion in the cerebellopontine angle without bone erosion. The patient refused surgery for three years and developed new symptoms due to trigeminal paresthesias, headache and dysphonia, as well as left peripheral facial paralysis. A suboccipital retrosigmoid approach was carried out, a pearly white lesion related to a cholesteatoma was found, confirmed by biopsy. Subtotal resection was achieved. He presented post-surgical chemical meningitis which resolved spontaneously. The evolution was satisfactory.
Conclusions: intracranial congenital cholesteatomas are very rare lesions and are sometimes similar to other lesions in the posterior cranial fossa. Imaging, they appear as well-defined lesions that do not present enhancement after contrast administration. In this patient, the evolution was favorable, despite the late surgical treatment, due to its initial refusal.
Keywords: MIDDLE EAR CHOLESTEATOMA/congenital; MIDDLE EAR CHOLESTEATOMA /surgery; DERMOID CYST; EPIDERMAL CYST; POSTERIOR CRANIAL FOSSA; EMISSION COMPUTED TOMOGRAPHY; MAGNETIC RESONANCE IMAGING; CASE REPORTS.
Recibido: 28/10/2021
Aprobado: 23/01/2022
INTRODUCCIÓN
Los colesteatomas o quistes epidermoides son lesiones benignas del oído medio, de crecimiento progresivo, consistentes en epitelio queratinizado que prolifera y sufre una descamación interna, responsable de su crecimiento, erosión ósea y compresión de estructuras adyacentes. Mientras los colesteatomas adquiridos aparecen como resultado de procesos inflamatorios crónicos en el oído medio, los colesteatomas congénitos se forman por el tejido epitelial embriológico residual, que puede aparecer en la porción petrosa, mastoidea o timpánica del hueso temporal.(1,2)
Los colesteatomas congénitos pueden alcanzar gran tamaño antes de ocasionar síntomas y revelan su presencia solo en estadios avanzados, cuando aparecen las complicaciones resultantes de hipertensión intracraneal o compresión de estructuras nerviosas. La definición de colesteatoma gigante se mantiene sin consenso, pero se acepta como una condición excepcional cuando sobrepasa los límites del hueso temporal y contacta con estructuras vitales como la arteria carótida interna y los nervios craneales, o con extensión intracraneal.(3)
La variante congénita es bastante rara con una incidencia anual de 0,12 por cada 100 000 personas.(4) En la mayoría de los casos, los síntomas son leves y afectan la esfera vestíbulo-coclear, con pérdida de la audición y desequilibrio, como los más frecuentes.(1) La intervención quirúrgica es la modalidad de tratamiento en todos los casos y la técnica a emplear depende de la extensión y localización de la lesión.(5) No existen reportes de casos similares en nuestro país.
Se presenta un paciente con diagnóstico de colesteatoma congénito de fosa posterior. Este artículo tiene como objetivo describir las características clínicas, imagenológicas, anatomopatológicas y la evolución postquirúrgica del mismo.
INFORMACIÓN DEL PACIENTE
Paciente masculino, de 48 años, de procedencia rural, obrero agrícola, con antecedentes de glaucoma y artritis reumatoidea desde hace 12 años. No se recogen antecedentes de enfermedades genéticas ni trastornos neoplásicos en familiares de primer grado de consanguinidad.
CUMPLIMIENTO DEL COMPONENTE ÉTICO DE LA INVESTIGACIÓN CLÍNICA
El comité de ética de la institución aceptó la publicación del informe de caso, previa aprobación del paciente. Se obtuvo la firma del paciente con el consentimiento informado para divulgar su situación de salud. Se mantuvo la observancia en la eliminación de la información identificativa de todos los datos relacionados con el paciente.
PERSPECTIVA DEL PACIENTE
El paciente se mantuvo cooperativo con la atención médica recibida. Los familiares y el paciente, al término del período de seguimiento, mostraron satisfacción con los resultados.
HALLAZGOS CLÍNICOS
El paciente comienza con tinnitus izquierdo, de aparición subaguda y evolución progresiva, descrito como un pitido intenso, con disminución de la audición. Luego de ocho semanas comienza a presentar pérdida del equilibrio, con caída a la izquierda durante la marcha con los ojos cerrados. Al examen físico se constató:
Sistema nervioso: Romberg simple con caída a la izquierda. Dismetría y discronometría ipsilateral. Nistagmo horizontal, con fase lenta hacia la izquierda. Weber lateralizado a la derecha. Rinne positivo anormal.
EVALUACIÓN DIAGNÓSTICA
Audiometría de tonos puros: hipoacusia sensorineural.
Se realiza tomografía axial computarizada (TAC) monocorte que informa tumoración, con diámetro máximo de 65 mm en sentido anteroposterior y 23 en lateralidad, hipodensa (6-10 UH) en el ángulo pontocerebeloso izquierdo con amplia comunicación, en sentido rostral, con cisternas prepontina, interpeduncular y ambiens en sentido caudal, hasta la cisterna magna. Se aprecian límites bien definidos, sin imágenes trabeculadas en su interior. Provoca un efecto de masa discreto sobre el tronco encefálico y el cuarto ventrículo, sin ocasionar hidrocefalia obstructiva. No se evidencian lesiones óseas; no existe captación del contraste yodado (Fig. 1).
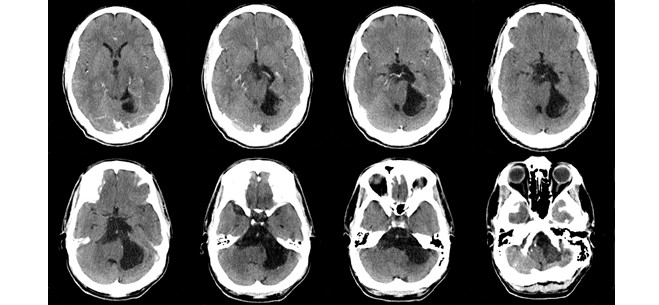
Fig. 1 - Imágenes de TAC monocorte con imagen tumoral a nivel del ángulo pontocerebeloso izquierdo con efecto de masa discreto sobre el tronco encefálico, sin captación de contraste yodado.
En la imagen de resonancia magnética nuclear se observó una lesión hiperintensa en T1 e hipointensa en T2, con imagen hetorogénea iso e hiperintensa en secuencia FLAIR (FLluid-Attenuated Inversion Recovery), lo cual excluyó la posibilidad inicial de quiste aracnoideo como diagnóstico presuntivo (Fig. 2).
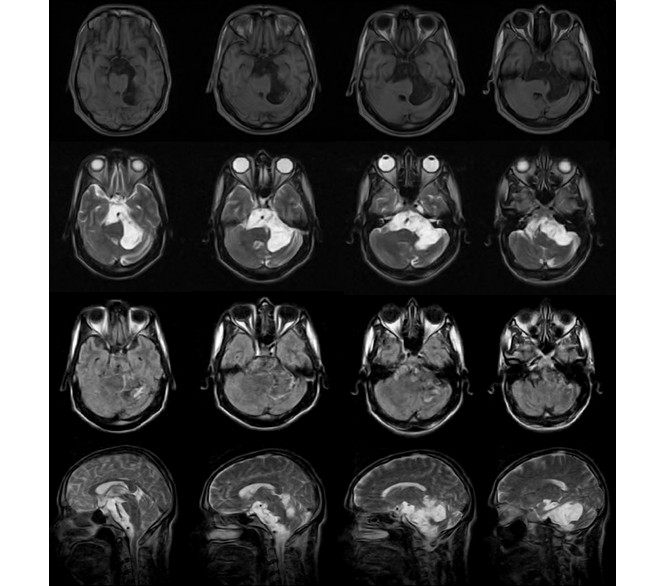
Fig. 2 - Imágenes de resonancia magnética nuclear.
INTERVENCIÓN TERAPÉUTICA
Se le plantea la posibilidad de tratamiento quirúrgico directo mediante abordaje retrosigmoideo suboccipital, para exéresis y biopsia, pero el paciente no da su consentimiento.
Luego de tres años sin evolución clínica y bajo seguimiento imagenológico anual, que no se reveló crecimiento de la lesión inicialmente descrita, comienza con parestesia facial, con distribución dermatómica en la segunda y tercera rama trigeminal izquierda, con sensación de hormigueo y ardor, e intensificación al cepillarse y durante la ingestión de alimentos. Además, aparece simultáneamente, cefalea suboccipital, moderada, opresiva, de carácter punzante, con predominio nocturno y, además, ligera disfonía.
El examen físico muestra parálisis facial periférica izquierda (Grado IV/VI según la escala de House-Brackman);(6) marcha con desviación a la izquierda, a la anulación visual; maniobra de Romberg simple con caída a la izquierda y nistagmo horizontal rotatorio. Además, se observa desviación de la úvula a la derecha y paresia de la cuerda vocal ipsilateral. Se constata hipoacusia perceptiva izquierda.
Previo consentimiento, se realiza craniectomía suboccipital retrosigmoidea y exéresis subtotal, técnica propuesta inicialmente, donde se constata una lesión blanca perlada (Fig. 3).

Fig. 3 - Imagen transoperatoria de la lesión con apariencia blanca perlada al examen macroscópico, en relación con colesteatoma.
En el informe de la biopsia se informan inclusiones de colesterol, compatible con un colesteatoma.
SEGUIMIENTO Y RESULTADOS
En el postoperatorio, presentó paresia del VI nervio craneal izquierdo, con recuperación total a las 48 horas. Al tercer día, comenzó con signos meníngeos y temperatura de 38,3 grados Celsius. Se le realizó punción lumbarpara estudios citoquímico y bacteriológico del líquido cerebroespinal, que informó resultados normales, sin evidencia de proceso infeccioso del sistema nervioso central, por lo que se interpretó como meningitis química post-quirúrgica, característica en este tipo de lesiones, la cual resolvió espontáneamente en un plazo de siete días.
El paciente ha mantenido una evolución favorable después de un año del tratamiento quirúrgico.
DISCUSIÓN
Los quistes epidermoides o colesteatomas se forman por descamaciones progresivas de derivados epiteliales.(1,2)
En la patogénesis de la variante congénita se acepta la inclusión de células ectodérmicas durante el cierre del tubo neural, entre la tercera y la cuarta semanas de vida intrauterina y la mayoría de los autores los consideran como malformaciones congénitas y no como verdaderas neoplasias.(7)
El trauma de la membrana timpánica por retracción, invaginación, perforación, desplazamiento, infecciones del oído, disfunción de la trompa de Eustaquio o enfermedades de la mucosa de la cavidad timpánica, tiene una relación causal con la variante adquirida.(8)
Los quistes epidermoides o colesteatomas representan entre 0,2 % y 1,8 % de los tumores intracraneales.(9) Muestran una incidencia similar en ambos sexos, cuando la población pediátrica y adulta son evaluadas en el mismo cohorte.(5,10)
La mayoría de los colesteatomas reportados son congénitos. Los síntomas pueden presentarse a cualquier edad, luego de largos períodos asintomáticos, independientemente de su origen congénito.(11) Los más frecuentes son otalgia, otorrea y disminución de la audición. Además, pueden causar abscesos y meningitis química aséptica por la ruptura del quiste en el espacio subaracnoideo, con la salida de queratina, colesterol y lípidos.(12)
El uso de la TAC y la imagen de resonancia magnética, es vital en el diagnóstico y la planificación quirúrgica.(1,13) La tomografía computarizada, por su accesibilidad, es la responsable del diagnóstico y la que informa sobre la dimensión y límites de la lesión. La apariencia típica es una masa extraaxial hipodensa y sin captación de contraste yodado. Han sido reportadas calcificaciones entre el 10 y 25 % de los casos.(14)
Las imágenes por resonancia magnética es el estudio imagenológico de elección. Las secuencias más usadas son las potenciadas en T1 y en T2. Los colesteatomas se muestran como iso o hipo-intensidades en T1 y ligeramente hiperintensos en T2, con una forma bien delimitada, ligeramente oval. El uso de nuevas secuencias de procesamiento como las imágenes por difusión (diffusion weighted imaging), marcan la diferencia en el proceso diagnóstico de estas lesiones, se muestran con alta hiperintensidad de señal debido a la restricción de la difusión de agua intracelular y por la consistencia grasa en la porción quística de la lesión dada la presencia de la queratina.(13,15)
El diagnóstico diferencial de estas lesiones debe realizarse con los quistes aracnoideos, quistes dermoides, neoplasias quísticas y quistes hidatídicos. Las imágenes por difusión diagnostican los quistes aracnoideos. Los dermoides muestran alta intensidad en imágenes potenciadas en T1. Los meduloblastomas quísticos y los ependimomas, generalmente poseen una apariencia heterogénea a causa de la hemorragia y necrosis y exhiben una captación heterogénea del contraste.(9)
El único tratamiento de estas lesiones es el quirúrgico, mediante la cual se logra su resección completa con la mínima morbilidad.(5,16) Los abordajes quirúrgicos estarán en relación con la localización del colesteatoma, la presencia de estructuras vitales como las arterias vertebrales y sus ramas o los senos venosos y el estado de la función auditiva. El apoyo microscópico o endoscópico permite la adecuada visualización de las estructuras neurales en la fosa posterior, lo que evita daños durante la intervención quirúrgica.(17,18)
En ocasiones se precisan abordajes combinados, en aras de lograr una mayor resección,(3) sin correr el riesgo de ocasionar un nuevo déficit o aumentar el preexistente, lo que deja en estos casos las porciones adherentes a estructuras vitales.(19,20) Como estas lesiones no muestran características típicas de una neoplasia, el pronóstico es bueno.(12)
Una de las limitaciones de este artículo, es el corto período de seguimiento de solo un año y la ausencia de imágenes de resonancia magnética por difusión, por razones logísticas.
CONCLUSIONES
Los colesteatomas congénitos intracraneales son lesiones muy infrecuentes. Se presentan generalmente con otalgia, otorrea y disminución de la audición. La resonancia magnética nuclear es vital para su diagnóstico; se presentan como lesiones hipo o isointensas en T1 e hiperintensas en T2, bien delimitadas, sin realce al contraste. En este caso la evolución ha sido favorable un año después del tratamiento quirúrgico, a pesar del retraso por la negativa inicial del paciente.
REFERENCIAS BIBLIOGRÁFICAS
- Davidoss N, Ha J, Banga R, Rajan G. Delayed Presentation of a Congenital Cholesteatoma in a 64-year-old Man: Case Report and Review of the Literature. J Neurol Surg Rep [Internet]. 2014 [citado 16 Abr 2020];75(1):e113-6. Disponible en: https://www.thieme-connect.de/products/ejournals/pdf/10.1055/s-0034-1376200.pdf
- Bharathi MB, Mehta P, Sivapuram K, Sandhya D. Cholesteatoma Classification: Review of Literature and Proposed Indian Classification System—TAMPFIC. Indian J Otolaryngol Head Neck Surg [Internet]. 2020 [citado 16 Abr 2020];14(1):402-9. Disponible en: https://link.springer.com/article/10.1007/s12070-020-02154-8
- Cazzador D, Favaretto N, Zanoletti E, Martini A. Combined Surgical Approach to Giant Cholesteatoma: A Case Report and Literature Review. Ann Otol Rhinol Laryngol [Internet]. 2016 [citado 16 Abr 2020];125(8):687-93. Disponible en: https://pubmed.ncbi.nlm.nih.gov/27117903/
- Tos M. A New Pathogenesis of Mesotympanic (Congenital) Cholesteatoma. Laryngoscope [Internet]. 2000 [citado 16 Abr 2020];110(11):1890-7. Disponible en: https://onlinelibrary.wiley.com/doi/epdf/10.1097/00005537-200011000-00023
- Misale P, Lepcha A. Congenital Cholesteatoma in Adults-Interesting Presentations and Management. Indian J Otolaryngol Head Neck Surg [Internet]. 2018 [citado 16 Abr 2020];70(4):578-82. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6224827/pdf/12070_2018_Article_1362.pdf
- House WE, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg. 1985;93(2):146-7.
- Niikawa S, Hara A, Zhang W, Sakai N, Yamada H, Shimokawa K. Proliferative assessment of craniopharyngioma and epidermoid by nucleolar organizer region staining. Childs Nerv Syst. 1992;8(8):453-6.
- Persaud R, Hajioff D, Trinidade A, Khemani S, Bhattacharyya MN, Papadimitriou N, et al. Evidence-based review of aetiopathogenic theories of congenital and acquired cholesteatoma. J Laryngol Otol [Internet]. 2007 [citado 16 Abr 2020];121(11):1013-9. Disponible en: https://www.cambridge.org/core/journals/journal-of-laryngology-and-otology/article/abs/evidencebased-review-of-aetiopathogenic-theories-of-congenital-and-acquired-cholesteatoma/65C0DF4A1D730938F06E2A0B4C703886
- Kumar S, Sharma S, Misra R, Kumar K. Epidermoid Cyst of the Fourth Ventricle: A Case Report. Indian J Neurosurg [Internet]. 2019 [citado 16 Abr 2020];08(3):191-2. Disponible en: https://www.thieme-connect.de/products/ejournals/pdf/10.1055/s-0039-1698843.pdf
- Danesi G, Cooper T, Panciera DT, Manni V, Côté DWJ. Sanna Classification and Prognosis of Cholesteatoma of the Petrous Part of the Temporal Bone: A Retrospective Series of 81 Patients. Otol Neurotol. [Internet]. 2016 [citado 16 Abr 2020];37(6):787–92. Disponible en: https://pubmed.ncbi.nlm.nih.gov/26808555/
- Maniu A, Harabagiu O, Perde Schrepler M, Catana A, Fanuta B, Mogoanta CA. Molecular biology of cholesteatoma. Roum J Morphol Embryol [Internet]. 2014 [citado 16 Abr 2020];55(1):7-13. Disponible en: http://www.rjme.ro/RJME/resources/files/550114007013.pdf
- Schwarz D, Gostian A-O, Shabli S, Wolber P, Hüttenbrink KB, Anagiotos A. Analysis of the dura involvement in cholesteatoma surgery. Auris Nasus Larynx [Internet]. 2018 [citado 16 Abr 2020];45(1):51-6. Disponible en: http://www.sciencedirect.com/science/article/pii/S0385814616304990
- Quan-Soon AY, Pulickal GG. Radiological Features of Acquired and Congenital Cholesteatoma. En: Pulickal GG, Tan TY, Chawla A, editores. Temporal Bone Imaging Made Easy. Cham: Springer International Publishing; 2021.
- Raghavan D, Lee TC, Curtin HD. Cholesterol Granuloma of the Petrous Apex: A 5-Year Review of Radiology Reports with Follow-Up of Progression and Treatment. J Neurol Surg B Skull Base [Internet]. 2015 [citado 16 Abr 2020];76(4):266-71. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4516729/
- Coman M, Coman A, Gheorghe DC. All about Imagistic Exploration in Cholesteatoma. Mædica [Internet]. 2015 [citado 16 Abr 2020];10(2):178-84. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5327812/pdf/maedica-10-178.pdf
- Sung CM, Yang HC, Cho YB, Jang CH. Congenital Cholesteatoma of Mastoid Temporal Bone and Posterior Cranial Fossa Treated with Transmastoid Marsupialization. Korean J Otorhinolaryngol-Head Neck Surg [Internet]. 2018 [citado 16 Abr 2020];61(12):710-3. Disponible en: https://synapse.koreamed.org/func/download.php?path=L2hvbWUvdmlydHVhbC9rYW1qZS9zeW5hcHNlL3VwbG9hZC9TeW5hcHNlWE1MLzAwMzhram9ybC9wZGYva2pvcmwtaG5zLTIwMTctMDAyOTcucGRm&filename=a2pvcmwtaG5zLTIwMTctMDAyOTcucGRm
- Iannella G, Savastano E, Pasquariello B, Re M, Magliulo G. Giant Petrous Bone Cholesteatoma: Combined Microscopic Surgery and an Adjuvant Endoscopic Approach. J Neurol Surg Rep [Internet]. 2016 [citado 16 Abr 2020];77(1):e46-9. Disponible en: https://www.thieme-connect.de/products/ejournals/pdf/10.1055/s-0035-1571205.pdf
- Dehadaray A, Kaushik M, Qadri H, Goyal P. Congenital cholesteatoma of petrous apex: Rare case report: Diagnostic and management challenge. Indian J Otol [Internet]. 2013 [citado 16 Abr 2020];19(2):75-8. Disponible en: https://www.indianjotol.org/article.asp?issn=0971-7749;year=2013;volume=19;issue=2;spage=75;epage=78;aulast=Dehadaray
- Nagasawa D, Yew A, Safaee M, Fong B, Gopen Q, Parsa AT, et al. Clinical characteristics and diagnostic imaging of epidermoid tumors. J Clin Neurosci [Internet]. 2011 [citado 16 Abr 2020];18(9):1158-62. Disponible en: http://www.sciencedirect.com/science/article/pii/S0967586811001263
- Bewarder J, Tóth M, Münscher A. Extensive cholesteatoma of the temporal bone with compression of the posterior fossa and midline displacement. Laryngo-Rhino-Otologie [Internet]. 2018 [citado 16 Abr 2020];97(s02):[aprox. 10 p.]. Disponible en: https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0038-1640260#htmlfulltext
Conflicto de intereses
Los autores declaran que no existen conflictos de intereses.
Contribución de los autores
Ernesto Enrique Horta-Tamayo: conceptualización, curación de datos, análisis formal, investigación, metodología y redacción-borrador original.
Carlos García-Alonso: curación de datos, investigación y recursos.
Luis César Acosta-González: análisis formal, investigación, metodología, recursos y redacción-revisión y edición.
Financiación
Hospital Clínico-Quirúrgico “Lucía Iñiguez Landín”.
