Publicación Trimestral de la Universidad de Ciencias Médicas de Ciego de Ávila Editorial Ciencias Médicas |
 |
REVISTA MÉDICA ELECTRÓNICA DE
CIEGO DE ÁVILA |
2020;26(4):e1806
ISSN: 1029-3035
RNPS: 1821 |
|

Presentación de caso
Síndrome de Peutz-Jeghers. Informe de caso
Peutz-Jeghers syndrome. Case report
Leidelen Esquivel-Sosa2* https://orcid.org/0000-0002-8062-8716
Yisel González-Ríos1 https://orcid.org/0000-0003-4833-6403
Manuel Lara-Martín3 https://orcid.org/0000-0002-8715-0241
1Máster en Ciencias. Especialista de Primer y Segundo Grados en Imagenología. Especialista de Primer Grado en Medicina General Integral. Profesor Auxiliar. Universidad de Ciencias Médicas de Villa Clara. Hospital Provincial Pediátrico Universitario “José Luis Miranda”. Villa Clara, Cuba.
2Especialista de Primer Grado en Medicina General Integral e Imagenología. Profesor Instructor. Universidad de Ciencias Médicas de Villa Clara. Hospital Provincial Pediátrico Universitario “José Luis Miranda”. Villa Clara, Cuba.
3Máster en Ciencias. Especialista de Primer y Segundo Grados en Gastroenterología. Especialista de Primer Grado en Medicina General Integral. Universidad de Ciencias Médicas de Villa Clara. Hospital Provincial Pediátrico Universitario “José Luis Miranda”. Villa Clara, Cuba.
*Autor para la correspondencia. Correo electrónico: leidelen@infomed.sld.cu
RESUMEN
Introducción: el síndrome de Peutz-Jeghers es una enfermedad rara, de herencia autosómica dominante. Se caracteriza por la aparición de máculas hiperpigmentadas en los labios, la mucosa oral y otras mucosas del organismo. Se asocia a poliposis hamartomatosa gastrointestinal.
Objetivo: presentar el caso de una paciente pediátrica con síndrome de Peutz-Jeghers, en cuyo diagnóstico fue fundamental el empleo de técnicas imagenológicas.
Presentación del caso: paciente femenina de seis años de edad. Presentó dolor abdominal recurrente, palidez cutáneomucosa, y lesiones maculares de color café en las mucosas de los labios superior e inferior y de la región peribucal. En los exámenes complementarios se le detectó anemia moderada. Se le realizaron exámenes imagenológicos, en los que observaron las asas intestinales engrosadas, fijas y con escasa perístasis. Por el estudio genético se confirmó que la paciente era positiva al síndrome de Peutz-Jeghers, y su padre portador.
Discusión: la incidencia de la enfermedad es de un caso entre 200 000. Su tasa de mortalidad es alta, pero la de prevalencia es baja. El diagnóstico se establece mediante cuatro criterios mayores; todos incluyen los pólipos, confirmados por examen histológico. La ecografía es fundamental para la detección y descripción de los engrosamientos parietales.
Conclusiones: la paciente reunió los criterios clínicos para la sospecha diagnóstica del síndrome de Peutz-Jeghers. Los estudios imagenológicos fueron fundamentales para establecer el diagnóstico, confirmado por el examen histológico y los estudios genéticos. La paciente no presentó neoplasias, debido a su corta edad. Por ello se le mantiene un seguimiento periódico en consulta.
Palabras clave: SÍNDROME DE PEUTZ-JEGHERS/diagnóstico; SÍNDROME DE PEUTZ-JEGHERS/diagnóstico por imagen; SÍNDROME DE PEUTZ-JEGHERS/cirugía; SÍNDROME DE PEUTZ-JEGHERS/genética; INFORMES DE CASOS.
ABSTRACT
Introduction: Peutz-Jeghers syndrome is a rare disease with autosomal dominant inheritance. It is characterized by the appearance of hyperpigmented macules on the lips, oral mucosa and other mucous membranes of the body. It is associated with gastrointestinal hamartomatous polyposis.
Objective: to present the case of a pediatric patient with Peutz-Jeghers syndrome, in whose diagnosis the use of imaging techniques was essential.
Case presentation: six-year-old female patient. She presented recurrent abdominal pain, mucous skin pallor, and brown macular lesions on the mucous membranes of the upper and lower lips and the perioral region. In the complementary examinations, moderate anemia was detected. Imaging examinations were carried out, in which they observed thickened, fixed intestinal loops with little perstasis. The genetic study confirmed that the patient was positive for Peutz-Jeghers syndrome, and her father was a carrier.
Disccusion: the incidence of the disease is one case in 200 000. Its mortality rate is high, but the prevalence rate is low. The diagnosis is established through four major criteria; all include polyps, confirmed by histological examination. Ultrasound is essential for the detection and description of parietal thickening.
Conclusions: the patient met the clinical criteria for the suspected diagnosis of Peutz-Jeghers syndrome. Imaging studies were essential to establish the diagnosis, confirmed by histological examination and genetic studies. The patient did not present neoplasms, due to her young age. For this reason, she is regularly monitored in consultation.
Keywords: PEUTZ-JEGHERS SYNDROME/diagnosis; PEUTZ-JEGHERS SYNDROME/diagnostic imaging; PEUTZ-JEGHERS SYNDROME/surgery; PEUTZ-JEGHERS SYNDROME/genetics; CASE REPORTS.
Recibido: 07/03/2020
Aprobado: 04/06/2020
INTRODUCCIÓN
Se han descrito múltiples síndromes en los cuales las enfermedades polipoideas intestinales se asocian a otras lesiones orgánicas. Entre ellos se encuentran los síndromes de Laugier-Hunziker, Carney, Cronkhite-Canada y Peutz-Jeghers. Este último aparece en pacientes de edades pediátricas.(1)
El síndrome de Peutz-Jeghers fue descrito por Jan Peutz en 1921, y en 1949 el médico norteamericano Harold Joseph Jeghers publicó de manera detallada 10 casos de pacientes con poliposis intestinal y pigmentación anómala de la piel; de ellos seis tenían antecedentes familiares. Ello permitió establecer que las causas del síndrome de Peutz-Jeghers son genéticas,(2) con herencia autosómica dominante debida a una mutación germinal del gen STK11/LKB1, localizado en el cromosoma 19p13. Esta enfermedad tiene una amplia heterogeneidad genética y penetrabilidad variable e incompleta.(3)
Este síndrome se caracteriza por la aparición de máculas hiperpigmentadas en los labios, la mucosa oral y otras mucosas del organismo. Esta sintomatología se asocia a poliposis hamartomatosa gastrointestinal, y los pacientes tienen elevada predisposición de presentar enfermedad neoplásica.(1,4,5) Su prevalencia alcanza hasta uno por cada 200 000 nacidos vivos, sin predominio de género ni raza. La edad promedio de diagnóstico comienza aproximadamente a los 9 años.(2,6)
Los pólipos con características histológicas típicas son más comunes en el intestino delgado, pero pueden aparecer en otras localizaciones como el estómago, colon y –menos frecuentemente– en el recto, además de vesícula biliar, bronquios, vejiga y uréter. Los pólipos intestinales causan dolor abdominal recurrente y vómitos.(1,4)
Durante muchos años no se realizaron estudios exploratorios del intestino delgado en estos casos. El desarrollo de nuevos medios diagnósticos ha facilitado el examen del tubo digestivo. Mediante la endoscopia, el ultrasonido, la tomografía axial computarizada y la resonancia magnética nuclear, es posible diagnosticar con certeza enfermedades inflamatorias y tumorales, entre otras, y evaluar las posibles terapéuticas a emplear.(7)
El objetivo de este trabajo es presentar el caso de una paciente pediátrica con síndrome de Peutz-Jeghers, en cuyo diagnóstico fue fundamental el empleo de técnicas imagenológicas.
PRESENTACIÓN DEL CASO
Paciente femenina de seis años de edad, sin antecedentes previos de enfermedad. Presentó dolor abdominal recurrente tipo cólico, de varias semanas de evolución, por lo cual fue atendida en su área de salud. Se le indicaron los exámenes complementarios de rigor, y se le detectó anemia moderada. Por este motivo fue ingresada en el Hospital Provincial Pediátrico Universitario “José Luis Miranda”, de Villa Clara. Por los exámenes complementarios que se le realizaron se corroboró que, además de la anemia, presentaba sangre oculta en heces fecales.
Examen físico:
La paciente presentó palidez cutáneomucosa, y lesiones maculares de color café, oscuras, en las mucosas de los labios superior e inferior y de la región peribucal (Fig.1).
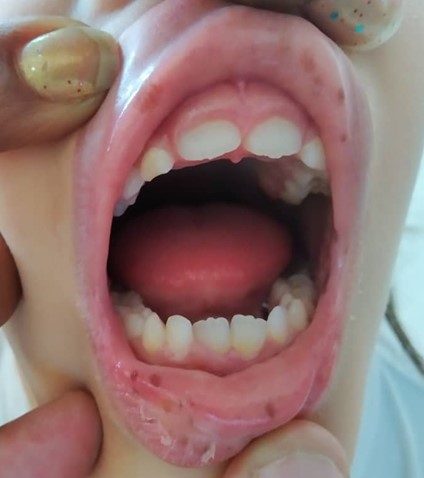
Fig. 1 - Máculas hiperpigmentadas en la mucosa labial.
Durante la palpación del abdomen se detectó que la paciente sufría un dolor difuso, sobre todo hacia el hipocondrio y el flanco izquierdo, sin reacción peritoneal.
Examen imagenológico:
Al segundo día de ingreso, como parte del estudio, se indicó la realización de un ultrasonido abdominal. En el cual llamaron la atención las asas intestinales engrosadas, localizadas en el flanco izquierdo, fijas y con escasa perístasis. También se observó que la luz de las asas estaba ocupada por una imagen ecogénica, de contorno lobulado y aspecto polipoideo. Se encontró otra imagen de asa, con anillos concéntricos, que recordaba una invaginación intestinal, movilizada durante el estudio (Fig. 2).
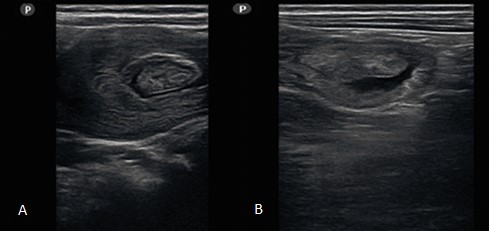
Fig. 2 - A. Se observa imagen en diana durante la invaginación. B. Imagen polipoidea en el interior del asa.
Ante este hallazgo, a la paciente se le realizó una tomografía axial computarizada contrastada de abdomen, en un tomógrafo multicorte, a 3 mm de espesor, con contraste yodado vías oral y endovenosa. Como resultados se informaron: múltiples asas yeyunales engrosadas (sin lograr adecuada repleción de contraste), y en el interior de su luz imágenes de defecto de llenado, con aspecto polipoideo, que no provocaban estenosis de la luz del asa (Fig. 3).
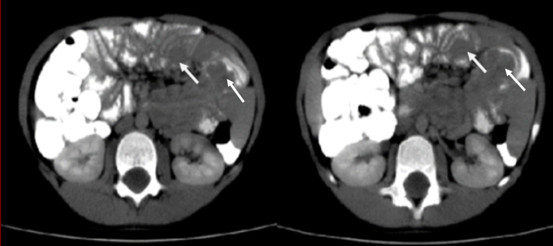
Fig. 3 - Asas con defecto de llenado e imágenes polipoideas en su interior.
En la interconsulta del caso con el especialista en gastroenterología, se indicó la realización de una endoscopia superior. En el procedimiento se demostró la presencia de pólipos de diferentes tamaños, visibles en el duodeno y la primera porción del yeyuno, hasta donde se logró introducir el endoscopio superior.
Dadas la rareza del caso y la existencia de una consulta especializada centralizada, se remitió a la paciente al Instituto Nacional de Gastroenterología para la confirmación diagnóstica del síndrome de Peutz-Jeghers. En la intervención quirúrgica realizada se efectuó resección de parte del intestino delgado, donde existían los mayores pólipos causantes del cuadro obstructivo. Se tomó muestra para biopsia, con resultado histológico de pólipos hamartomatosos.
En estudio genético se confirmó que la paciente era positiva al síndrome de Peutz-Jeghers, y su padre portador de la enfermedad. Actualmente se encuentra en seguimiento por personal altamente capacitado, sin nuevas complicaciones.
DISCUSIÓN
El síndrome de Peutz-Jeghers es de causa genética y se encuentra dentro del grupo de enfermedades clasificadas como raras. Su incidencia es de un caso entre 200 000, con alta tasa de mortalidad y baja prevalencia.(1-2)
En 1895, ante la Sociedad Científica de Londres, el doctor JRT Conner describió por primera vez este síndrome, en unas gemelas idénticas de 12 años de edad, que presentaban pigmentación en los labios y la boca. Una murió a los 20 años por obstrucción intestinal, y la segunda a los 52 por cáncer de mama. En 1921 el médico holandés Jan Peutz describió siete miembros de una familia holandesa que presentaban pigmentación mucocutánea y poliposis en intestinos y nasofaringes. En 1924 Van Dijk y Oudenal describieron un caso similar de dos hermanos. Posteriormente Jeghers y colaboradores (1949) describieron 10 casos de diferentes familias y publicaron las características del síndrome. El epónimo “de Peutz-Jeghers” fue otorgado por Bruwer y colaboradores, de la Clínica Mayo, en el año 1954.(2,3)
Este síndrome es de transmisión autosómica dominante, con penetrancia incompleta. Su causa generalmente es una mutación en la línea germinal del gen supresor de tumores STK11/LKB1, localizado en el cromosoma 19p13. Los pacientes con truncamiento del gen en lugar de una mutación se afectan de forma mucho más grave, lo cual sugiere una correlación del fenotipo y el genotipo. Casi la mitad de los pacientes se deben a nuevas mutaciones.(1-4)
El síndrome de Peutz-Jeghers se caracteriza por la aparición de máculas hiperpigmentadas, típicamente en el borde de los labios, la mucosa oral, y otras mucosas como encías y ano, así como en la piel de manos y pies. Las lesiones cutáneas crecen en tamaño y número hasta la pubertad, después de lo cual comienzan a remitir, aunque las máculas pigmentadas bucales tienden a persistir. Los enfermos presentan, además, hamartomas del tubo digestivo, localizados principalmente en el intestino delgado, pero también en el colon, el estómago y, con menor frecuencia, en el recto. En los niños a menudo se evidencia el exceso de estrógenos, causante de ginecomastia y edad ósea avanzada.(1-5)
En comparación con la población general, los enfermos del síndrome de Peutz-Jeghers tienen entre 10 y 18 veces mayor riesgo de presentar enfermedades neoplásicas. De ellas la de mayor morbilidad es la neoplasia maligna gastrointestinal, 130 veces más frecuente en pacientes con síndrome de Peutz-Jeghers. La localización de estas neoplasias es variable: colon (39 %), estómago (29 %), e intestino delgado (13 %). No obstante, pueden aparecer también en otros órganos: especialmente en las mamas, el tracto genitourinario, las gónadas y el páncreas (en esta última localización el riesgo es 100 veces mayor en comparación con la población general).(1-4)
Las neoplasias comienzan a aparecer alrededor de los 30 años de vida. Por el alto riesgo de estos enfermos de padecer cáncer, se recomienda aplicarles protocolos de detección estándar. Debido a que 40 % desarrollan complicaciones gastrointestinales significativas y, potencialmente, cáncer genitourinario a los seis años, es posible que la detección gastrointestinal deba comenzar tan pronto como a los cuatro o cinco años.(1-4)
Para el diagnóstico del síndrome de Peutz-Jeghers existen cuatro criterios mayores: presencia de dos o más pólipos confirmados histológicamente; cualquier número de pólipos característicos del síndrome, asociados a historia familiar de la enfermedad; pigmentación mucocutánea característica y un familiar con síndrome de Peutz-Jeghers y existencia de cualquier cantidad de pólipos característicos del síndrome, asociados a pigmentación mucocutánea.(1,4)
Se debe tener en cuenta que la poliposis del intestino delgado puede causar episodios repetidos de dolor abdominal y vómitos. Los pólipos grandes pueden infartar, ulcerarse y sangrar, así como causar invaginación y obstrucción intestinal(4,5), como ocurrió la paciente cuyo caso se expone aquí.
El diagnóstico diferencial se debe establecer con los síndromes de Laugier-Hunziker, de Carney de Cronkhite-Canadá, dado que todos ellos se caracterizan por manifestaciones cutáneas y poliposis gastrointestinal, pero con características diferentes en cada uno. También difieren en cuanto a la edad de aparición, pues estos síndromes aparecen en pacientes mayores de 30 años. Los enfermos del síndrome de Carney también pueden desarrollar tumores de células de Sertoli y ginecomastia. Este último detalle, en combinación con la pigmentación mucocutánea característica, puede conducir a un diagnóstico erróneo de síndrome de Peutz-Jeghers.(1)
En los últimos 20 años se han conseguido progresos considerables en el diagnóstico del síndrome de Peutz-Jeghers, sobre todo en cuanto al examen del segmento del intestino delgado. Años atrás se estudiaba el tránsito intestinal, pero actualmente se indica poco porque solo permite definir la luz intestinal o –en menor medida, y con el empleo de la técnica de doble contraste– la superficie mucosa, de forma indirecta. Su rendimiento diagnóstico es limitado, e incluye el uso de radiación ionizante, comparable a la de una enterotomografía.(7)
Entre las técnicas empleadas en la actualidad se destaca la ecografía, muy útil para detectar y describir los engrosamientos parietales, cuando las condiciones de ecogenicidad son buenas. La tomografía axial computarizada con contraste endovenoso y la enterotomografía con enteroclisis permiten explorar de forma relativamente exhaustiva todo el intestino delgado, si la distensión es buena en toda su longitud. La enterotomografía también permite explorar el resto de la cavidad abdominopélvica, sobre todo el mesenterio, el peritoneo y los ejes vasculares. Sin embargo, este examen implica emisión de rayos X, por lo cual no es adecuado para el seguimiento regular de enfermedades crónicas.(7)
Más recientemente se ha incorporado la enterorresonancia magnética. Esta es una exploración sin radiaciones ionizantes, por lo cual puede repetirse sin riesgo y es muy adecuada para el estudio inicial y el seguimiento de enfermedades crónicas intestinales. Pero, en comparación con la enterotomografía, la eficacia de la enterorresonancia magnética es menor en la detección de lesiones tumorales u otras anomalías morfológicas. Ello se debe a que la distensión del intestino delgado suele ser menor, al igual que la resolución espacial de esta técnica que, además, es muy sensible a los artefactos producidos por el movimiento intestinal.(7)
Para realizar la endoscopia se utilizan diversas vías. La menos frecuentemente usada es la enteroscopia peroperatoria. No obstante, se indica en casos raros como las polipectomías múltiples en enfermos del síndrome de Peutz-Jeghers. La finalidad de esta elección es evitar resecciones reiteradas del intestino delgado, que podrían causar un síndrome de intestino corto.(7-9)
El tratamiento de estos pacientes consiste en el seguimiento regular de su estado gastrointestinal. De esta forma se verifica la formación de pólipos y se realiza la resección oportuna de los más grandes, con lo cual se previenen complicaciones y reducen los riesgos de transformación maligna.(5)
Las medidas de cribado de los pacientes del síndrome de Peutz-Jeghers deberían incluir la exploración de los testículos, del tracto gastrointestinal (mediante endoscopia gastroduodenal, colonoscopia, tránsito intestinal o cápsula endoscópica), la mamografía y las ultrasonografías abdominal, ginecológica y endoscópica pancreática, o resonancia magnética nuclear. La detección de cáncer de mama, ginecológico y testicular debe ser de rutina después de los 18 años.(8,9)
Sin embargo, ningún protocolo de cribado ha sido validado en ensayos clínicos. Existe una guía de recomendaciones, que incluyen no solo la vigilancia endoscópica de pólipos gastrointestinales, sino también el cribado de neoplasias extraintestinales como el cáncer de mama o testicular. Recientemente, el grupo internacional para el cribado de cáncer de páncreas incluyó dentro de los pacientes con alto riesgo a los enfermos de síndrome de Peutz-Jeghers, independientemente de la historia familiar de cáncer de páncreas.(10)
Este trabajo presentó como limitación la escasa literatura sobre el tema, debido a la rareza de síndrome, lo cual no permitió cotejar el caso con otros.
CONCLUSIONES
La paciente reunió los criterios clínicos establecidos por la Organización Mundial de la Salud para la sospecha diagnóstica del síndrome de Peutz-Jeghers. Como aporte de este trabajo, se subraya que los hallazgos de los estudios imagenológicos fueron fundamentales para establecer el diagnóstico. Este fue confirmado por el examen histológico y los estudios genéticos de la paciente, que resultaron positivos al síndrome de Peutz-Jeghers en su caso, mientras se identificó al padre como portador. Aunque por lo general la enfermedad evoluciona desfavorablemente por el riesgo elevado de aparición de neoplasias, la paciente no las presentó aún debido a su corta edad. Por ello se le mantiene un seguimiento periódico en consulta.
REFERENCIAS BIBLIOGRÁFICAS
- James WD, Elston DM, Treat JR, Rosenbach MA, Neuhaus IM. Disturbances of pigmentation. En: James WD, Elston DM, Treat JR, Rosenbach MA, Neuhaus IM. Andrews' diseases of the skin. Clinical dermatology. 13ra ed. Edinburgh: Elsevier; 2020. p. 862-80.
- Coronado AK, Chanis-Águila R. Síndrome de Peutz-Jeghers como causa de invaginación intestinal en niños. Pediátr Panamá [Internet]. Dic 2018 [citado 3 May 2020];47(3):24-8. Disponible en: http://docs.bvsalud.org/biblioref/2019/02/980130/12.pdf
- Rodríguez-Lagos FA, Sorlí-Guerola JV, Romero-Martínez IM, Codoñer-Franch P. Registro y seguimiento clínico de pacientes con síndrome de Peutz Jeghers en Valencia. Rev Gastroenterol Méx [Internet]. Jun 2020 [citado 3 Jun 2020];85(2):123-39. Disponible en: https://reader.elsevier.com/reader/sd/pii/S0375090619300485?token=02D96BF436AF34EB6DC8CFD1EFE30FC26C38383629FD4346C6D223274DA6E868906789441F951A7F45B2E9FA1609EBC8&originRegion=us-east-1&originCreation=20210409143958
- Aguilera-Matos I, Díaz-Oliva SE, Velazco-Villaurrutia YC, García-Bacallao E, Labrada-Moreno LM. Síndrome de Peutz-Jeghers, experiencia de casos en el Instituto de Gastroenterología. Arch cuba gastroenterol [Internet]. 2020 [citado 3 May 2020];1(1):e18. Disponible en: http://revgastro.sld.cu/index.php/gast/article/download/18/47
- McGarrity TJ, Amos CI, Baker MJ. Peutz-Jeghers syndrome. En: Adam MP, Ardinger HH, Pagon RA, Wallace SE. Bean LJH, Mirzaa G, editores. GeneReviews [Internet]. Seattle: University of Washington; 2000 [citado 20 May 2020]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1266/
- Roesch-Dietlen FB, Cano-Contreras AD, Meixueiro-Daza A, Remes-Troche JM, Grube-Pagola P. Obstrucción intestinal por cápsula endoscópica en un paciente con síndrome de Peutz-Jeghers. Rev Gastroenterol Méx [Internet]. Jun 2018 [citado 3 May 2020];83(2):202-4. Disponible en: https://www.sciencedirect.com/science/article/pii/S0375090617300307?via%3Dihub
- Saurin JC, Muller A, Benech N. Exploración del intestino delgado: principios e indicaciones. EMC-Tratado Medicina [Internet]. Mar 2017 [citado May 2020];21(1):1-6. Disponible en: https://doi.org/10.1016/S1636-5410(16)81793-6
- Cubiella J, Marzo-Castillejo M, Mascort-Roca JJ, Amador-Romero FJ, Bellas-Beceiro B, Clofent-Vilaplana J, et al. Guía de práctica clínica. Diagnóstico y prevención del cáncer colorrectal. Actualización 2018. Gastroenterol Hepatol [Internet]. Nov 2018 [citado 3 May 2020];41(9):585-96. Disponible en: http://www.alianzaprevencioncolon.es/imagenesAdmin/articulos/Gu%C3%ADa%20de%20pr%C3%A1ctica%20cl%C3%ADnica.%20Diagn%C3%B3stico%20y%20prevenci%C3%B3n%20del%20c%C3%A1ncer%20colorrectal.%20Actualizaci%C3%B3n%202018.pdf
- Wendel D, Murray KF. Tumores del tubo digestivo. En: Kliegman R, Stanton B, St Geme J, Schor NF, Behrman RE, Wilson KM. Nelson. Tratado de Pediatría. 21ra ed [Internet]. Barcelona: Elsevier; 2020. p. 2061-64. Disponible en: https://www.clinicalkey.es/#!/content/book/3-s2.0-B9788491136842003721
- Adán-Merino L, Aldeguer-Martínez M, Lozano-Maya M, Hernández-García-Gallardo D, Casado-Fariña I. Luces y sombras en el diagnóstico y seguimiento de un paciente joven asintomático con síndrome de Peutz-Jeghers. Rev Gastroenterol Méx [Internet]. Mar 2016 [citado May 2020];81(1):59-61. Disponible en: http://www.revistagastroenterologiamexico.org/index.php?p=revista&tipo=pdf-simple&pii=S037509061500066X
Conflictos de intereses
Los autores declaran que no existen conflictos de intereses.
Contribuciones de los autores
Leidelén Esquivel-Sosa: atención al paciente, idea original del artículo, búsqueda y análisis bibliográfico, redacción del manuscrito y aprobación de la versión final.
Yisel González-Ríos: atención al paciente, idea original del artículo, búsqueda y análisis bibliográfico, redacción del manuscrito y aprobación de la versión final.
Manuel Lara-Martín: atención al paciente, idea original del artículo, búsqueda y análisis bibliográfico, redacción del manuscrito y aprobación de la versión final.
Financiación
Hospital Provincial Pediátrico Universitario “José Luis Miranda”.
